Dimethyl Sulfoxide Oxidations
Dimethyl sulfoxide-based oxidation of primary alcohols to aldehydes or secondary alcohols to ketones is a mild method that does not depend upon heavy metal oxidants. Here is a brief, chronological history of the procedure.
Index:
-
Swern Oxidation (trifluoroacetic anhydride)
-
Swern Oxidation (oxalyl chloride)
Kornblum Oxidation: (1959) A primary tosylate is heated at 150o to cause SN2 displacement by the oxygen of dimethyl sulfoxide (DMSO) in the presence of NaHCO3. The accepted mechanism at the time was an E2 elimination as shown. Dimethyl sulfide (DMS) is the reduction product of the reaction. This work formed the tosylate from the alkyl iodide with silver tosylate. Presumably the tosylate can be prepared from the alcohol which is a more direct route. The downside of the procedure is the elevated temperature although the contact time is only a few minutes. The essential issue is to find a method to generate the sulfenate salt.

Barton Modification: (1964) Barton and coworkers were able to generate sulfenate salts by treating alkyl chloroformates with DMSO with loss of CO2. Addition of triethylamine generates the oxidation product. This procedure ameliorated the harsh conditions of the Kornblum procedure. Moreover, the chloroformate is readily available by treatment of the alcohol to be oxidized with excess phosgene.

Moffatt-Pfitzner Oxidation: (1963) This variant of a dimethyl sulfoxide-based oxidation was developed at Syntex. The activator is prepared by protonation of dicyclohexyl carbodiimide (1, DCC), which is attacked by dimethyl sulfoxide. Intermediate 2 is again protonated to facilitate addition of the alcohol oxygen on the sulfur atom. Stable dicyclohexyl urea 4 is formed along with sulfenate salt 3. This species suffers collapse to the carbonyl compound under the influence of the dihydrogenphosphate anion. Although phosphoric acid is an effective acid catalyst for this reaction, sulfuric acid, hydrogen chloride and trifluoroacetic acid are not. However, pyridinium trifluoroacetate is an effective catalyst. It is critical that the conjugate base of the acid is basic enough to effect the last step of the reaction.

Torrsell Mechanism: (1966) The Swedish chemist Torrsell demonstrated that the mechanism of these DMSO-based oxidations do not proceed via an intermolecular β-elimination. The elimination is an intramolecular process. Sulfenate salt 5 was prepared by the Moffatt-Pfitzner procedure (see above). If the Kornblum mechanism were to apply, the dimethyl sulfide obtained would be devoid of deuterium (Experiment 1). However, dimethyl sulfide-d1 (7) was obtained. This result is consistent with a base that deprotonates the methyl group attached to sulfur forming ylid 6. The ylid decomposes by intramolecular deprotonation to produce the observed products.
Experiment 1:

To corroborate this mechanism the deuterium was incorporated into DMSO as DMSO-d6. Accordingly, the reaction produced dimethyl sulfide-d5(12) and undeuterated aldehyde 11 (Experiment 2).
Experiment 2:

Parikh-Doering Oxidation: (1967) This oxidation utilizes the pyridine sulfur trioxide complex (13) as the activator of dimethyl sulfoxide. Sulfate is the leaving group in the displacement by the alcohol (primary or secondary) in intermediate 14. Sulfenate 15 is decomposed by the intramolecular mechanism (vide supra) to yield an aldehyde or ketone.

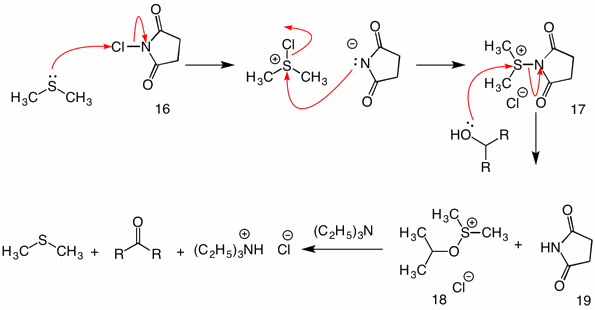
Corey-Kim Oxidation: (1972) In this procedure dimethyl sulfide is activated (oxidized) with N-chlorosuccinimide (16) to provide reagent 17. In the reaction of this species with an alcohol, the succinimidyl group functions as a leaving group. The usual sulfenate intermediate 18 collapses by the intramolecular mechanism upon the addition of triethylamine.
Swern Oxidation: (1976) This early Swern oxidation employs trifluoroacetic anhydride (20) at -50oC to activate dimethyl sulfoxide. Addition of the alcohol to intermediate 21 yields the desired sulfenate 22. The ketone or aldehyde is produced in the usual fashion with triethylamine.

Swern Oxidation: (1978) This later Swern procedure is a convenient method for the production of reagent 24 without using dimethyl sulfide and chlorine. Dimethyl sulfoxide, which is at the same oxidation level as salt 24, reacts with oxalyl chloride (23) to liberate carbon monoxide, carbon dioxide and reagent 24. Addition of the primary or secondary alcohol followed by deprotonation of sulfenate 25 with triethylamine leads to the desired aldehyde or ketone, respectively.
