Facial, Enantiotopic and Diastereotopic Descriptors
Index:
Trigonal Atom Xabc : Facial Descriptors
Diastereotopic Atoms, Enantiotopic Groups
Trigonal Atom Xabc : Facial Descriptors
The descriptors R and S are used most commonly to describe the chirality of asymmetrically substituted carbon atoms. There are instances where the faces of achiral molecules need to be distinguished. In these cases the terms Re and Si are employed, derived from the sam roots as R (L., rectus: right, in the sense of being correct) and S for lefthanded (L.,sinister: improper). Here is the paper.
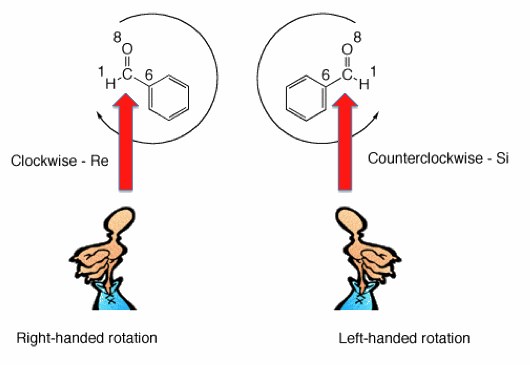
Consider the planar molecule benzaldehyde shown below. The carbon of the carbonyl group has a hydrogen, carbon (as phenyl) and oxygen attached to it in a trigonal arrangement. The faces of this molecule are said to be enantiotopic because any achiral nucleophile that is not H, phenyl or OH leads to two enantiomers. These three atoms are assigned their atomic numbers by the CIP convention. If they are arranged in descending order (8 --> 6 --> 1) in a clockwise fashion, the face is defined as Re. If the arrangement is counterclockwise, the designation is Si. You can easily use your hand to make the assignment in a similar way that we apply your hands to R,S notation. Point your thumb toward the face in question. If you rotate about the axis of your thumb and the remaining fingers rotate in the direction (8 --> 6 --> 1), then the face is defined by the hand employed. Benzaldehyde is straight forward because all three atoms have different atomic numbers.
 |
 |
|---|
| Consider C2 in the face of propene shown in 1a. Its three attached atoms (ligands) are C, C and H. How does one distinguish between the two carbons? Using the Cahn-Ingold-Prelog (CIP) method, the C-C double bond is treated as a single bond with "phantom" carbons attached to each of the carbons of the double bond (1b). Each of the ligand atoms of C2 are in red. Diagram 1c distinguishes the two carbon atoms (see blue atoms) while the "phantom" carbon at C2 is dropped, since this carbon must be trigonal. Now C1 [6(6,1,1)] has priority over C3 [6(1,1,1)]. The face of propene in 1a is Si. Clearly, C1 in 2a cannot be classified as Re or Si on the basis of the attached ligands: C, H, H. Dropping the phantom carbon on C1 in 2b accomplishes nothing; the priorities are still C, H, H. By convention, C1 is assigned as Si by virtue of the Si assignment at C2. | 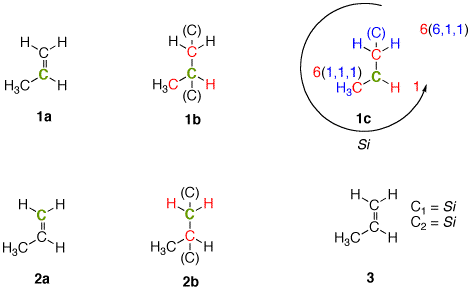 |
|---|
| These rules for facial descriptors of trigonal atom X with ligands A, B and C are summarized by the diagram on the right with priority decreasing in the order A > B > C. | 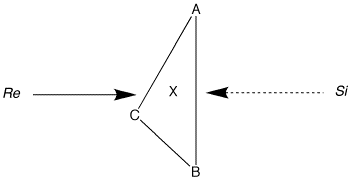 |
|---|
|
Homotopic atoms or groups possess an n-fold (n = 2, 3, ...) axis of symmetry. Rotation of methyl chloride (on the right) about the C-Cl axis has a 3-fold axis of rotation. The hydrogens are said to be homotopic. Another way to view homotopicity is that the atoms or groups are indistinguishable. Place your cursor over the hydrogens in methyl chloride. They are labeled H3, H4 and H5. Click your mouse button and rotate the structure rapidly so that you loose track of which hydrogen is which. Can you identify H3 by just looking at the three hydrogens. No you cannot! |
|---|
|
The methyl hydrogens of ethanol are homotopic but the hydrogens of the methylene group are heterotopic, i.e., different (H6 and H7; see above). In this instance they are enantiotopic. If you pass a plane through the two carbons and the hydroxyl group, the two hydrogens of the methylene group occupy enantiomeric positions. One is on the left; the other on the right. It is possible to distinguish these two hydrogens after rotating the molecule rapidly. These enantiotopic hydrogens bare the descriptors pro-R and pro-S. The assignment is made by substituting one of the hydrogens with deuterium and assigning the R/S configuration. If the R-configuration is obtained; the exchanged hydrogen is pro-R. H6 is pro-R; H7 is pro-S. |
|---|
|
The methylene hydrogens [H7 (green) and H8 (blue)] in (R)-2-bromobutane are heterotopic and diastereotopic owing to the stereogenic center. Substituting either one with deuterium leads to a different diastereomer. Substituting H8 for deuterium gives (2R,3S)-2-bromo-3-deutereobutane. H7 affords (2R,3R)-2-bromo-3-deutereobutane. H7 is pro-R; H8 is pro-S. The mirror image argument is equally applicable to (S)-2-bromobutane. |
|
|---|
Diastereotopic Atoms, Enantiotopic Groups:
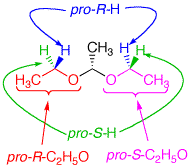
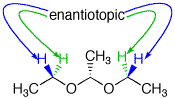
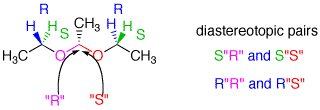
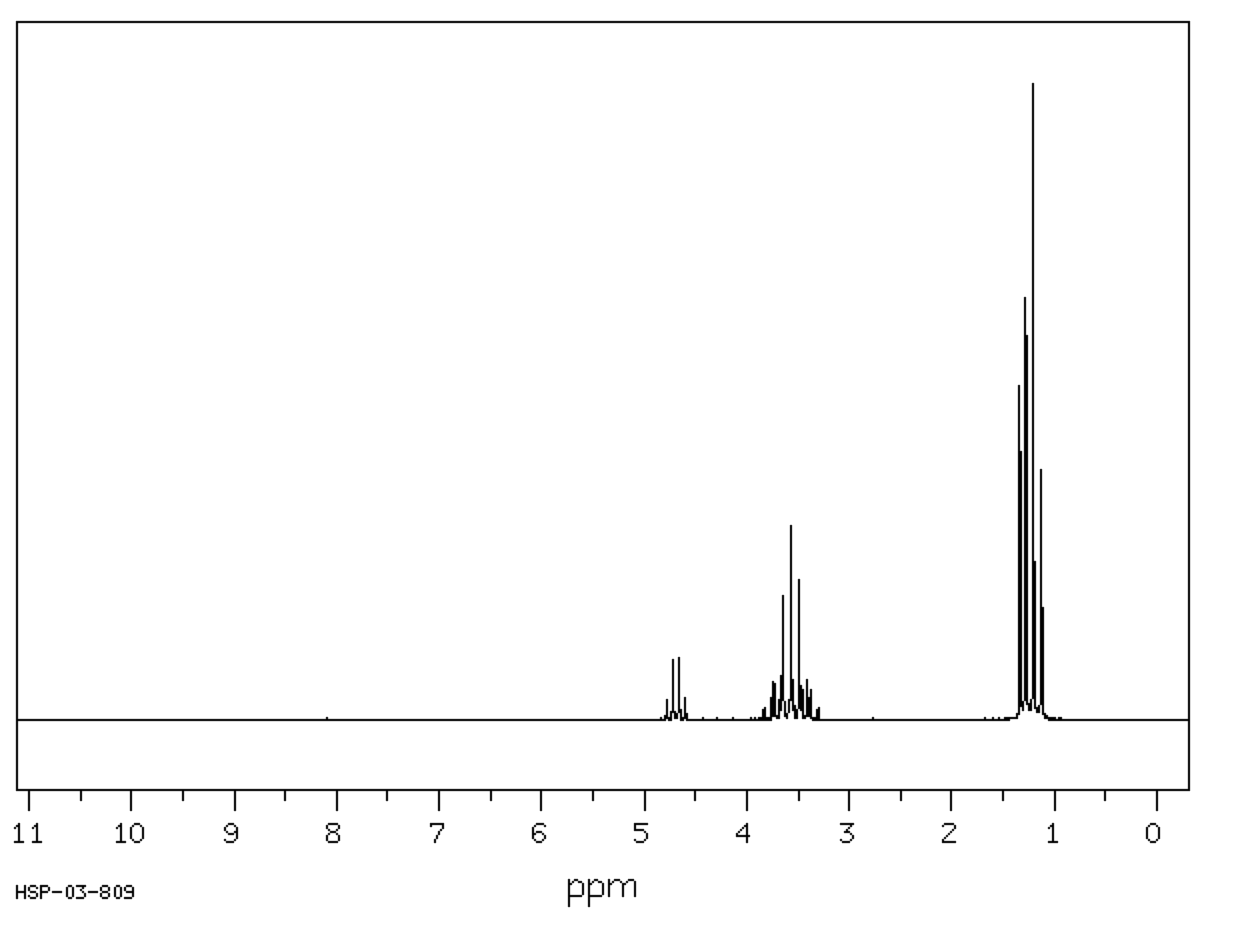
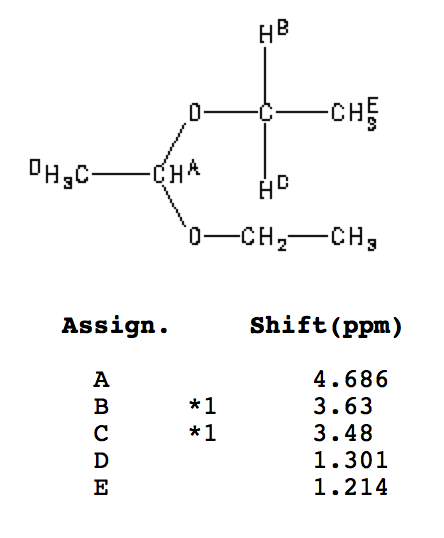
The diethyl acetal of acetaldehyde is an achiral molecule. In fig. 1 and the JSmol structure (below) both blue hydrogens are pro-R while the green hydrogens are pro-S. The ethoxy groups are enantiotopic. Fig. 2 illustrates enantiotopic pairs of hydrogens. There is a vertical (yz-plane) mirror plane reflecting each side of the molecule. Fig. 3 illustrates that the blue and green hydrogens are pairs of diastereotopic hydrogens. As determined in fig. 1, the pro-R hydrogens leads to R-configurations; the pro-S hydrogens afford an S-configuration. Consider a chemical gremlin sitting on the lefthand blue hydrogen. He sees an R-configuration at the methylene to which his hydrogen is attached. In addition, isotopic substitution at the magenta oxygen (O-18) gives the gremlin a view of the center carbon as "R". His view may be described as R"R". When our gremlin sits on the righthand blue hydrogen he still has an R-configuration at the carbon to which his hydrogen is attached but a mirror image view (red oxygen; O-18) of the central carbon as "S". This new view is R"S". The relationship R"R"/R"S" is diastereomeric. To put the argument another way, 180o rotation about the axis bearing the center carbon and its contiguous methyl group does superimpose the blue hydrogens but now the vicinal methyl group and hydrogen on the central carbon have switched positions. This operation makes the two blue hydrogens distinguishable. They are diastereotopic atoms. Test this statement with the Jmol structure. A similar argument can be made for the green hydrogens. The diastereomeric nature of the methylene protons in acetaldehyde diethyl acetal are clearly revealed in the 90 MHz 1sup>H NMR [fig.4; courtesy of SDBSWeb: http://sdbs.db.aist.go.jp (National Institute of Advanced Industrial Science and Technology), 6/2/2014]. The methylene protons at δ3.63 and δ3.48 are coupled to each other and the methyl group. This area of the spectrum is more complex than a simple quartet. The two signals overlap at 90MHz. |
fig. 1
fig. 2
fig. 3
fig. 4
|
|---|