Index:
Oxidations and reductions are commonly achieved by reagents that are electron-deficient or electron-rich, respectively. Typically, CrO3-based reagents convert primary alcohols to aldehydes and carboxylic acids and secondary alcohols to ketones. Each of these oxidation products can be reduced with LiAlH4 to their respective alcohols. Another way to effect oxidations and reductions is by redistributing electrons within a structure with reagents that are neither oxidants nor reductants. The classical Wolff-Kishner reaction is a case in point (Scheme 1). An aldehyde or ketone, in this instance cyclohexanone (1), is derivatized as its hydrazone 2 with hydrazine. In the presence of a strong base in ethylene glycol at elevated temperature (Huang Minlon variation, 1946) the hydrazone is deprotonated. The resonance stabilized anion 3 is protonated on carbon affording the the diimide 4. What has happened at this point is a two electron reduction of the carbon framework and a two electron oxidation of the dinitrogen moiety. Deprotonation of diimide 4 affords anion 5 that now adds another two electrons to the carbon moiety with a concommitent loss of two electrons from the diimide. Protonation of anion 6 (there is still debate as to whether a highly basic anion exists as such in this medium) provides the reduction product. The role of the base is to shuffle electrons from the nitrogen region of the molecule to the carbon region. In essence, hydrazine loses four electrons to give nitrogen gas while cyclohexanone gains four electrons to afford the product of reduction cyclohexane.
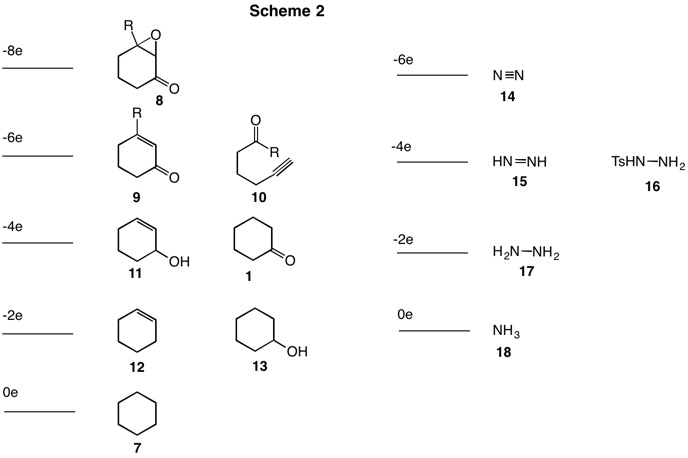
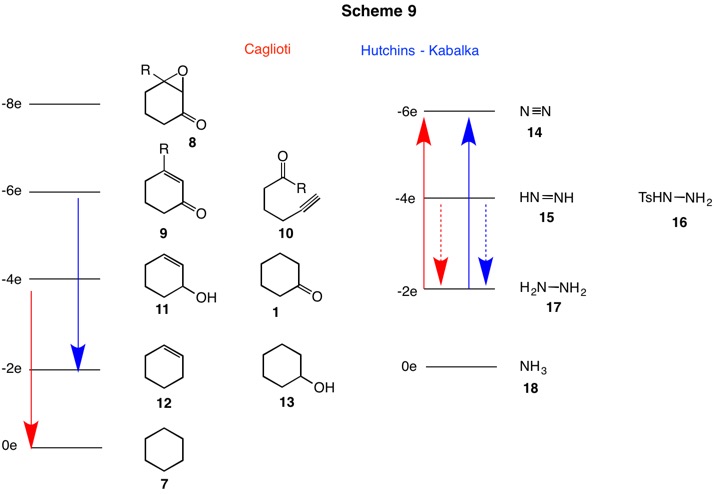
Before proceeding with the Wolff-Kishner story, consider the information in Scheme 2. The cyclohexane derivatives run from cyclohexane (7, arbitrarily set at 0 electrons) to the epoxyketone 8 (-8e level; R=H for this discussion). Each level has two fewer electrons than the level below it. Clearly, cyclohexene (12) and cyclohexanol (13) are interconvertible by hydration/dehydration, a procedure that does not involve oxidation or reduction. [For a review of oxidation levels, click here.] On the right side of Scheme 2 is illustrated the successive oxidation products of ammonia: ammonia (18), hydrazine (17), diimide (15) and nitrogen (14). Compound 16 is p-toluenesulfonylhydrazine (tosylhydrazine). It is prepared from p-toluenesulfonyl chloride (TsCl) and hydrazine. Elimination of TsH from 16 gives diimide and p-toluenesulfinic acid. The process is a two electron reduction of the tosyl group (Ts+ ---> Ts-).
Now we are in a position to combine the information in Schemes 1 and 2 (Scheme 3) to give a new paradigm for the Wolff-Kishner (W-K) reaction (red arrows) and related hydrazine-based reactions. [A slight digression. The use of the term W-K reduction is correct in that the focus in organic chemistry is on the carbon framework. But if you were asked for a method to oxidize hydrazine to nitrogen, would you have thought of the W-K reaction?] There is an overall gain of four electrons for the carbon framework and a loss of four electrons for the nitrogen moiety. The cyclohexyldiimide 4 has a cyclohexane ring that is at the oxidation level of cyclohexanol (13; +2e) while the nitrogenous portion of this intermediate is at the oxidation level of diimide (15; -2e).

Now you should be able to predict the type of structure that will form when the tosyl (p-toluenesulfonyl, Ts) hydrazine (16) derivative of cyclohexanone is treated with base. One need not know the actual mechanism but one has to presume that nitrogen gas is evolved. The oxidation now involves a two electron loss (blue arrows). Therefore, the reduction must involve a two electron gain. The anticipated product is either an alkene 12 or a substitution product as in alcohol 13. The Bamford-Stevens and Shapiro reactions meet this goal. However, the mechanisms of the two reactions are quite different.
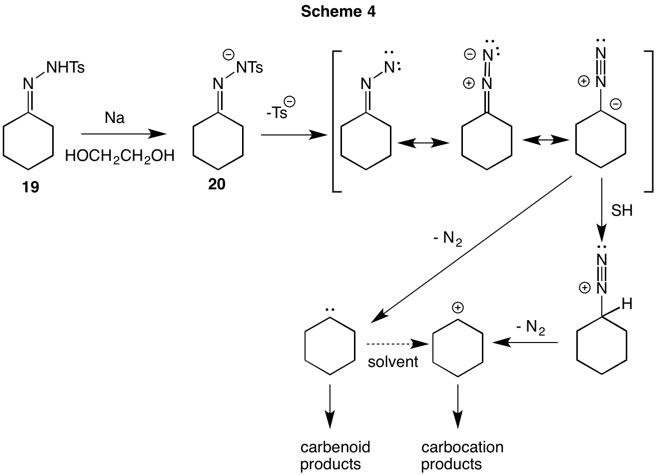
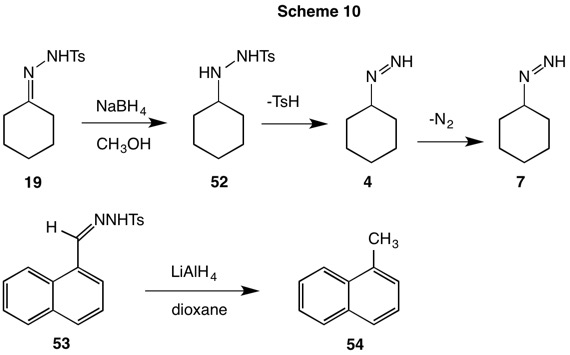
The Bamford-Stevens reaction (1952, Scheme 4) of aldehyde or ketone tosylhydrazones was historically conducted with sodium dissolved in ethylene glycol at elevated temperatures. Facile deprotonation of tosylhydrazone 19 affords anion 20, which undergoes α-elimination to give a neutral, resonance stable diazoalkane. In protic solvents carbocation and elimination products often form while in aprotic solvent carbenoid products are typical. The carbenoid pathway leads to alkenes and C-H insertion products. In the case of the cyclohexane tosylhydrazone 19, cyclohexene is formed by rearrangement of the carbene. Powell and Whiting (1959) demonstrated that the unimolecular loss of p-toluenesufinate anion from anion 20 was the rate limiting step in the formation of the secondary diazo compound. Although Bamford and Stevens reported quantitative formation of cyclohexene in 1952, Powell and Whiting were able to isolate the carbocationic product, β-cyclohexyloxyethanol in 20-35% yield
The Shapiro reaction (1967, Scheme 5) employs 2 equivalents of alkyllithium to form the dianion 21. The dianion undergoes elimination of the tosylsulfinate anion (24) to produce the vinyldiimide anion 22 which in turn loses nitrogen affording vinyllithium 23. Under these conditions the vinyllithium 23 does not survive because the ortho hydrogens of the tosylsulfinate anion 24 are sufficiently acidic to protoate the anion. Thus, trapping of the vinyllithium with electrophiles is prohibited.
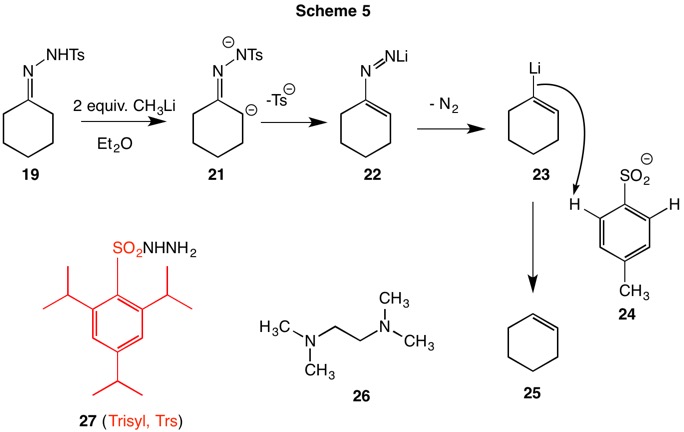
One solution to this problem was devised by Bond (1975, Scheme 5). Phenylhydrazones were employed to preclude the possibility that the methyl group of the tosylsulfinate anion could be a proton source and to employ four equivalents of butyllithium which not only formed the dianion but also, presumably, deprotonated the phenylsulfinate at the ortho positions. The vinylanion is also capable of effecting β-elimination of the solvent, diethyl ether. To avoid the latter problem, the lithium chelating solvent tetramethylethylenediamine (26, TMEDA) was employed. Only the efficiency of deuteration was explored. In 1978 Chamberlain and Bond introduced 2,4,6-trisopropylphenylsulfonylhydrazine (27) as a derivatizing agent. The lack of ortho hydrogens on the sulfinate anion (in red) precluded deprotonaton and hexane/TMEDA sufficed as a solvent system. Moreover, collapse of the vinylazo anion was more rapid than in the tosyl series. The vinyllithium was successfully trapped with a range of electrophiles.
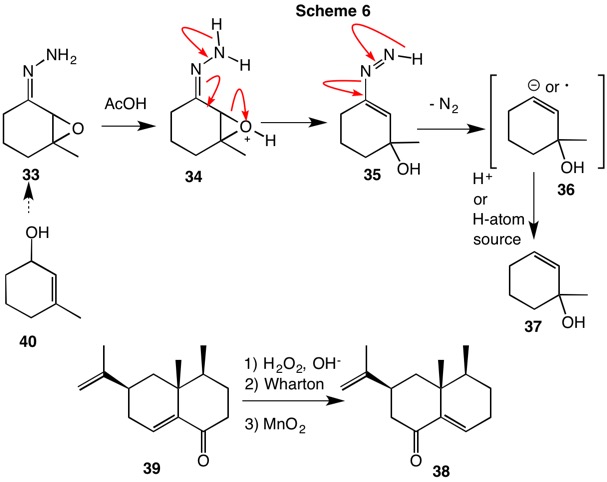
The Wharton Reaction (1961, Scheme 6) involves the decomposition of the hydrazone of an α,β-epoxyketone. The hydrazine loses four electrons in forming nitrogen. Therefore, the epoxyketone must gain four electrons (Scheme 3) and reach the oxidation level of a ketone or an allylic alcohol. The reaction of hydrazone 33 affords the allylic alcohol 37. Like the Wolff-Kishner reaction, there is evidence that radicals as opposed to carbanions are involved in the decomposition of the vinyldiimide 35. The process can be utilized for the inversion of allylic alcohols. Oxidation, epoxidation and hydrazone formation converts allylic alcohol 40 to 37. Historically, the reaction was catalyzed by acetic acid but bases can be employed in the transformation. The sesquiterpene eremophilone 38 has been prepared from the transposed enone 39 using the Wharton reaction.
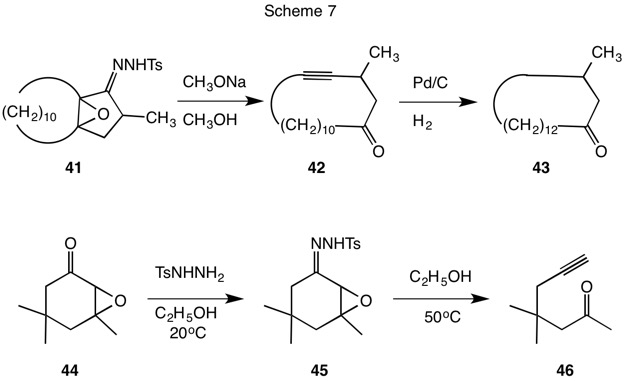
In an effort to develop a method for the preparation of the perfume ingredient muscone (43), Eschenmoser (ETH) and Ohloff (Firmenich) reported (1967, Scheme 7) the fragmentation of the tosylhydrazone 41 in the presence of base formed the cyclic acetylenic ketone 42. Hydrogenation produced racemic muscone (43). The facility of the fragmentation was exemplified when the process was shown to be initiated by silica gel. In the same year, Tanabe (SRI) published the same fragmentation. Isophorone epoxide 44 formed the unstable tosylhydrazone 45. Simply heating the ethanolic solution of the tosylhydrazone 45 at 50oC gave rapid evolution of nitrogen with concommitent formation of the acetylenic ketone 46. The analogous reaction with cyclohexene epoxide failed to give the acetylenic aldehyde. Referring to Scheme 3 once again, the conversion of tosylhydrazine to nitrogen is a two electron oxidation. Therefore, a two electron reduction of the epoxy ketone 8 is required. Likely products may include an unasturated ketone 9 or, as has been witnessed, an acetylenic ketone 10.
Two closely related mechanisms (Scheme 8) have been proposed for the reaction. Base can deprotonate the tosylhydrazone 47 and the resultant anion opens the epoxide in a vinylogous fashion. Collapse of vinyldiimide 48 via a doubly vinylogous Grob fragmentation leads the acetylenic ketone 49. Alternatively, α-elimination of p-toluenesulfinic acid from epoxide 47 can form the diazo epoxide 50, which fragments via the vinyl diazonium species 51.

Variations on a Theme: Caglioti (1966)
The reactions that have been discussed thusfar involve derivatizing carbonyls of different oxidation levels with hydrazine and its tosyl derivative -- both of different oxidation levels -- and to subsequently relocate electrons under the aegis of acid or base. No additional reductants or oxidants have been explored. The strongly alkaline conditions and elevated temperatures of the traditional Wolff-Kishner reaction engendered interest in developing milder methods for effecting this four electron reduction. Rather than employing hydrazine (17) to generate the diimide in the traditional way, tosylhydrazine was utilized in its stead. Hydride reduction of the tosylhydrazone double bond and elimination of tosylsulfinate would lead to the correct diimide intermediate. This was the approach explored by Caglioti (Scheme 9, red arrows) using NaBH4 or LiAlH4. The dotted red arrow of Scheme 9 brings the tosylhydrazone of cyclohexanone (19) to the oxidation level of the hydrazone. Under controlled conditions reduction product 52 could be isolated in 60-70% yield. Heating this intermediate in refluxing methanol quantitatively produced cyclohexane, nitrogen and p-toluenesulfinic acid (Scheme 10). Similarly, the tosylhydrazone 53 afforded α-methylnaphthalene in 70% yield.
 |
 |
|---|
Variations on a Theme: Hutchins
In the 1970's Hutchins investigated the reduction of tosylhydrazones with sodium cyanoborohydride (NaCNBH3) rather than with sodium borohydride (NaBH4). Unlike NaBH4, NaCNBH3 is stable in mildly acidic medium. Because aldehydes and ketones are reduced much slower than protonated imines --or, tosylhydrazones -- the tosylhydrazone may be generated in the presence of tosylhydrazine, NaCNBH3, the carbonyl compound and the acid catalyst, p-toluenesulfonic acid. The reaction was conduct at ~100oC in a mixed solvent system of dimethylformamide (DMF) and sulfolane [(CH2CH2)2SO2]. Unhindered aliphatic ketones were readily reduced by this procedure but hindered aliphatic ketones, such as 3,3,5-trimethylcyclohexanone, required preformed tosylhydrazone prior to reduction. Aromatic aldehydes and ketones were not effectively reduced by these techniques.
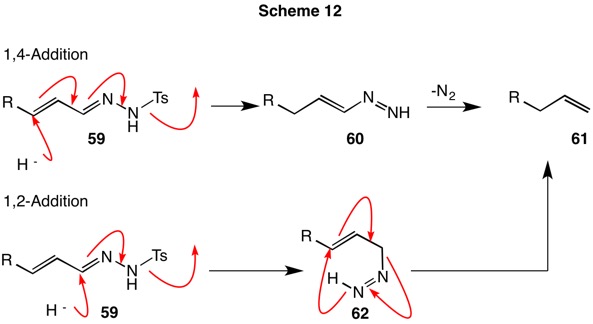
Reduction of α,β-unsaturated ketone tosylhydrazones with NaCNBH3 produced an alkene (see Scheme 9, blue arrows.). Surprisingly, the double bond was located between the α-carbon and the carbon of the former carbonyl group. The cyclohexenyl methyl ketone tosylhydrazone 55 provided ethylidene cyclohexane 56. Similarly, the tosylhydrazone of cinnamaldehyde 57 gave rise to 3-phenylpropene (58) (Scheme 11). Two possible mechanisms were considered (Scheme 12). Hydride delivery in a 1,4-fashion to conjugated tosylhydrazone 59 leads to vinyl diimide 60, which collapses to the alkene 61. Alternatively, hydride delivery in the 1,2-mode results in allylic diimide 62. Rearrangement gives rise to the observed product 61.
 |
 |
|---|
| Gribble demonstrated that sodium borohydride in acetic acid (HOAc) and tetrahydrofuran (THF) served as an effective substitute for sodium cyanoborohydride. Using this reagent protocol, Hutchins investigated the reduction of β-ionone tosylhydrazone (63, Scheme 13) with the goal of resolving the mechanism of the reduction of conjugated substrates. Reduction of tosylhydrazone 63 with sodium borodeuteride (NaBD4) in HOAc/THF incorporated a single vinylic deuterium atom at the carbon that had been part of the carbonyl group. This result confirms 1,2-reduction. The hydrogen of the diimide, or one that is exchanged with acetic acid, is delivered to the β-carbon via a pericyclic reaction. Secondly, NaBH4/DOAc delivered hydride to the vinylic site and deuterium to the β-carbon in diene 65. The latter reaction also showed that deuterium exchanges with tosylhydrazone 63 or the intermediate diazene did occur. In addition, when NaBD4/DOAc was employed, both carbons were deuterated. | 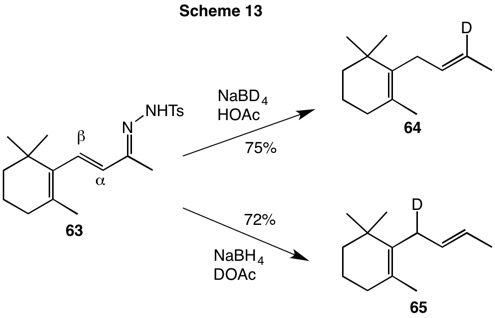 |
|---|
Variations on a Theme: Kabalka
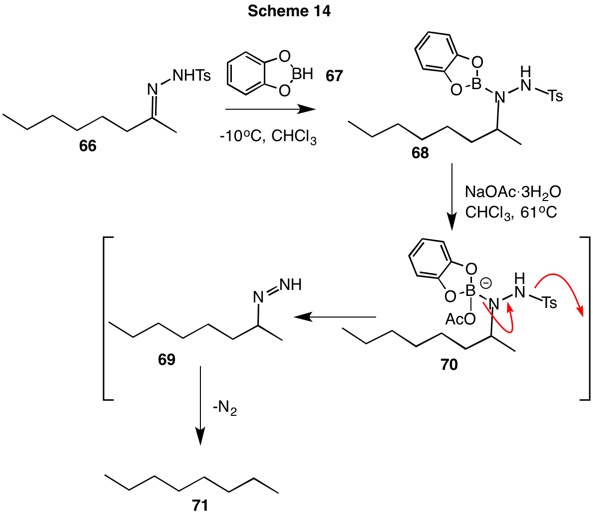
Kabalka discovered that the use of catecholborane (67, Scheme 14) had advantages over the conditions of Hutchins (vide supra): a) only a 10% excess of catecholborane was needed as opposed to a four-fold excess of NaCNBH3; b) low boiling point chloroform could be used instead of the higher boiling DMF-sulfolane solvent system; c) mild temperatures. Treatment of the tosylhydrazone of 2-octanone (66) with catecholborane led to the reduction product 68 at low temperature. The use of sodium acetate trihydrate permitted nucleophilic decomposition of 68 by acetate anion while the water served as the proton source for the decompositionn of alkyl diimide 69 to n-octane 70.
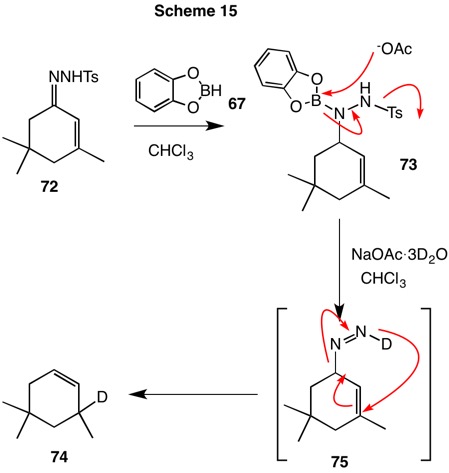
This methodology was effective in the reduction of the tosylhydrazone of isophorone (72, Scheme 15). Sodium acetate hydrated with 3 equivalents of D2O allows H/D exchange at either reduction product 73 or allylic diimide 75. The rearrangement of the diimide requires the azo group to be axially oriented in the transition state. As previously shown in the studies of Hutchins, the double bond migrates in the reduction and deuterium is incorporated to afford cyclohexene 74.
 |
 |
|---|
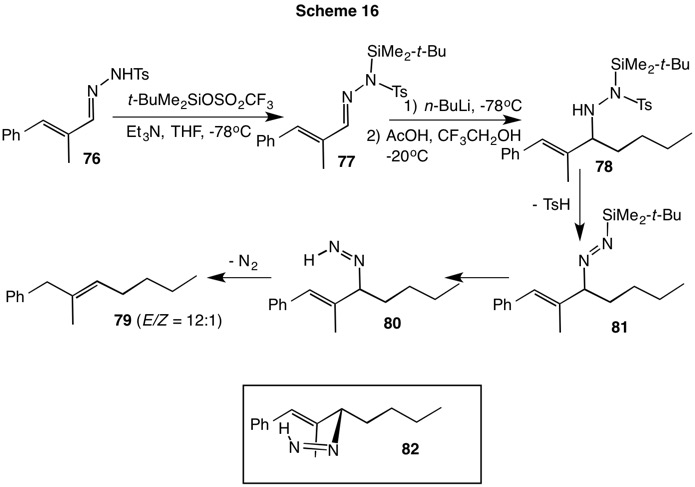
The work of Hutchins and Kabalka focused on the reduction of ketone tosylhydrazones to produce the labile alkyl tosylhydrazines. Myers approached this goal by adding alkyllithium reagents to aldehyde tosylhydrazones to achieve the same end. The use of basic alkyllithium reagents required substitution of the tosylhydrazone's acidic hydrogen with a silane protecting group lest Bamford-Stevens or Shapiro reactions occur. In Scheme 16 conjugated tosylhydrazone 76 is first protected with tert-butyldimethylsilyl triflate at low temperature to produce the derivatized tosylhydrazone 77. Exposure of the protected tosylhydrazone to n-butyllithium at low temperature affords the lithium amide of 78. Addition of acetic acid and β,β,β-trifluoroethanol at -20oC initiates a cascade of reactions. Loss of p-toluenesulfinic acid from 78 afforded the silyldiimide 81. Desilylation of 81 led to the diimide 80, which rearranged with double bond migration and loss of nitrogen. The predominate (E)-isomer arises owing to the n-butyl group being equatorial in the transition state 82.
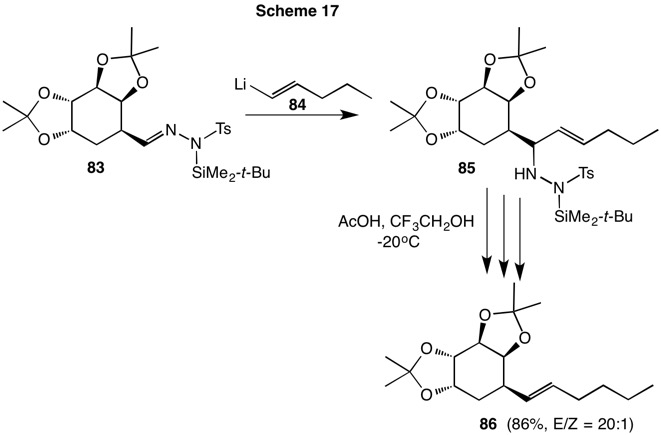
In another example (Scheme 17), the silyl tosylhydrazone 83 was not conjugated while the (E)-alkenyllithium 84 provided the source of the double bond. Decomposition of adduct 85 led to a migrated (E)-double bond with higher selectivity than in the case of alkene 79. This is because the bulk of the bis-acetonide moiety of alkene 86 is much larger in the transition state for rearrangement than the n-butyl group of dimide 80..
 |
 |
|---|
Variations on a Theme: Yamamoto, The Catalytic Shapiro Reaction
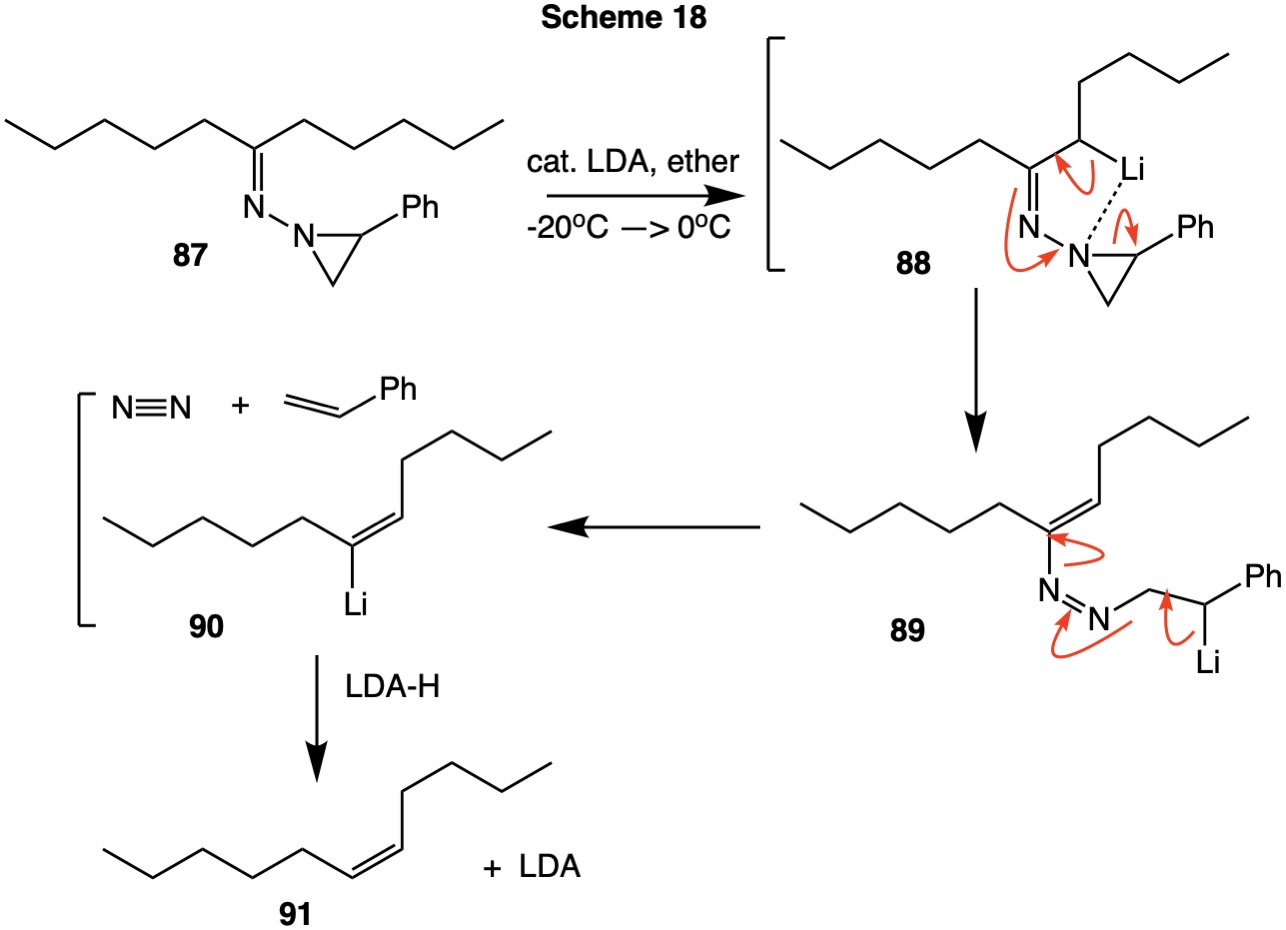
Derivatization of the symmetrical ketone 6-undecanone with 2-phenylaziridin-1-amine forms the hydrazone 87 (Scheme 18). Treatment of the hydrazone with a catalytic amount of lithium diiisopropylamide (LDA) forms the enolate 88 that is chelated to the aziridine ring. Fragmentation of this species leads to benzylic anion 89 with a (E)-configuration of the C-C double bond cisoid as to the carbon framework. Collapse of 89 liberates the (Z)-organolithium 90 in addition to styrene and nitrogen. The configurationally stable organolithium reagent is protonated by diisopropylamine (LDA-H) to produce (Z)-5-undecene with the regeneration of LDA.
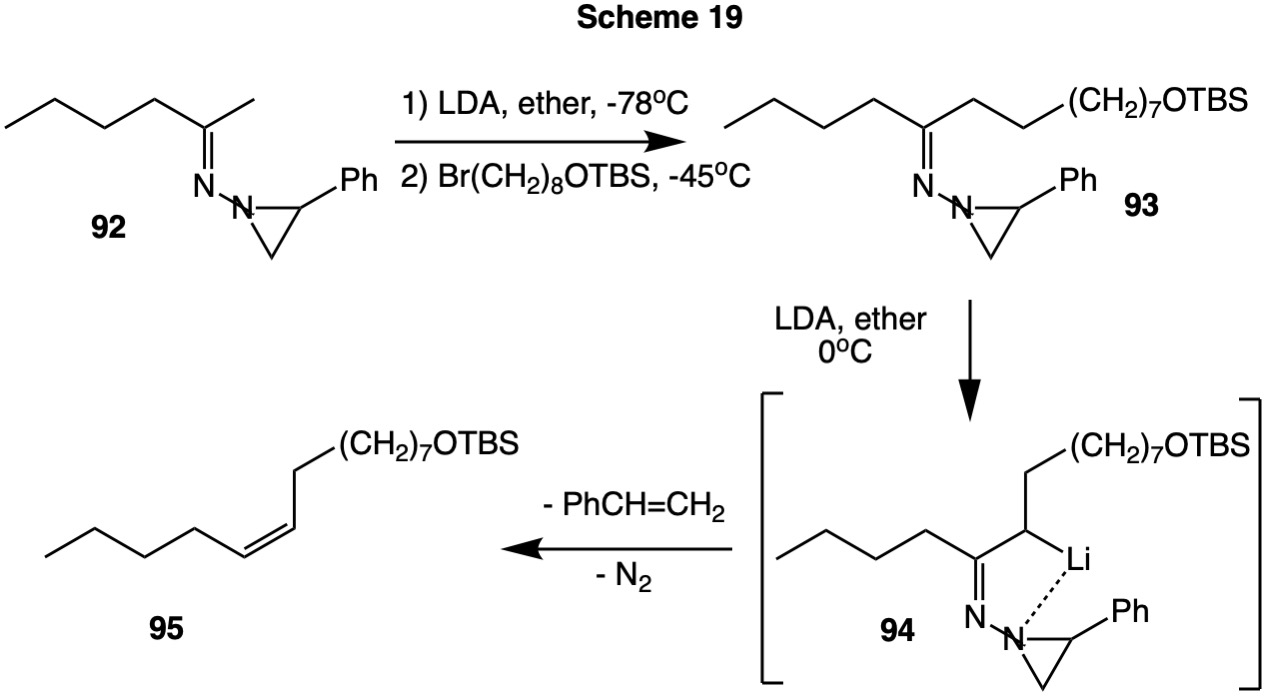
The (E)-hydrazone 92 formed from 2-hexanone can be deprotonated on the methyl group at -78oC with a small excess LDA without fragmentation. Alkylation affords the (Z)-hydrazone 93, which, upon raising the temperature to 0oC, regioselectively deprotonates syn to the aziridine ring and undergoes fragmentation to (Z)-alkene 95.
 |
 |
| When the achiral ketone 4-methyl-4-(p-tolyl)cyclohexan-1-one is derivatized with chiral (R)-2-phenylaziridin-1-amine, two diastereomers are formed: (R,R)-96 and (R,S)-99. In both instances, the aziridine ring controls the site of deprotonation. Each diastereomer produces its own chiral enantiomer. Thus, (R,R)-96 affords (R)-cyclohexene 98 while (R,S)-99 gives (S)-cyclohexene 101. | 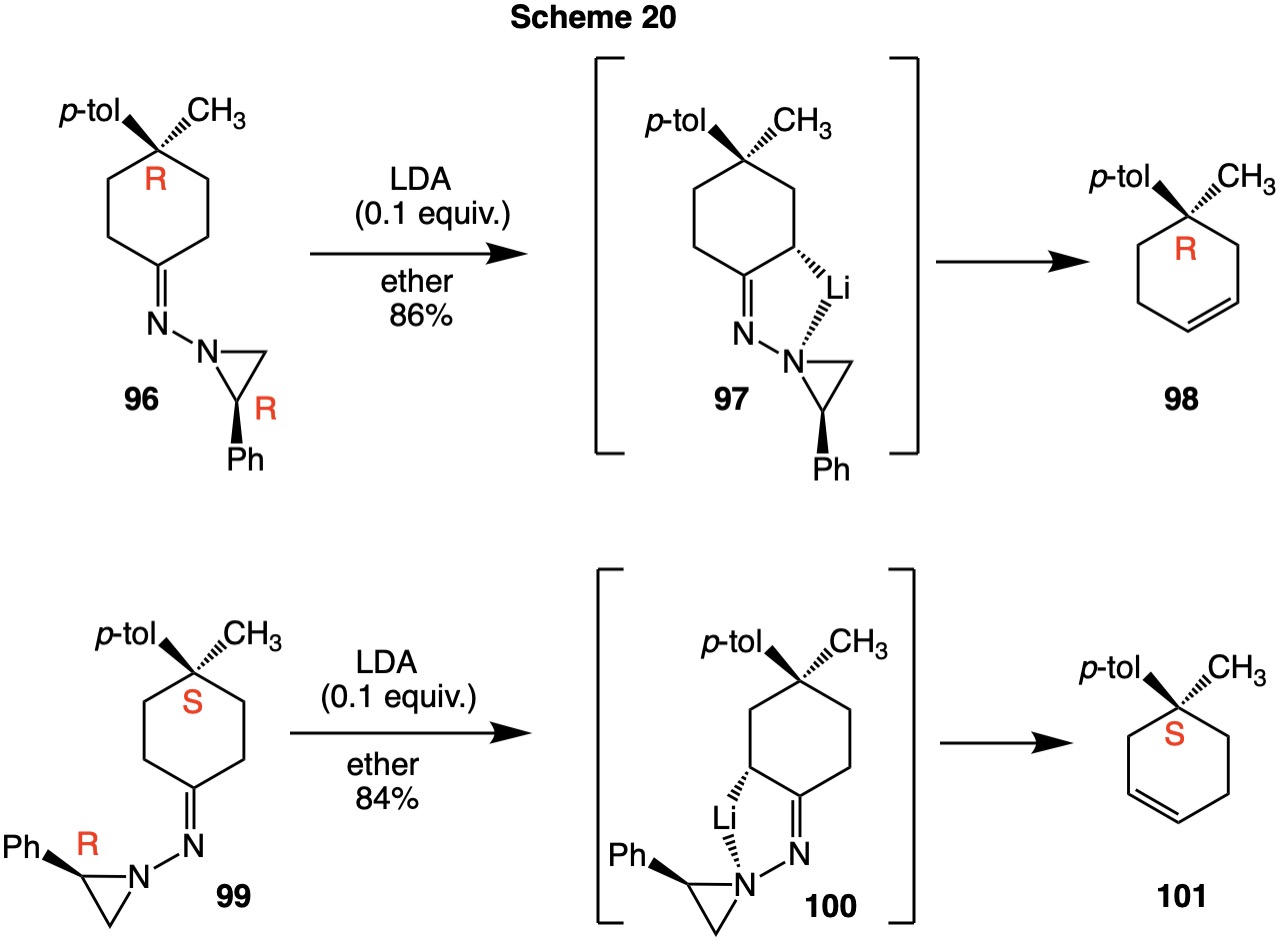 |
F. E. Ziegler 9/22/2016; expanded 10/12/2016