 Leif D.
Jacobson
Leif D.
Jacobson
 Leif D.
Jacobson
Leif D.
Jacobson
| Research Interests Publications Software Contributions Recreational Interests Education Contact Homepage |
My
interests are in the fields of electronic structure theory and chemical
dynamics. I am particulary interested in the interface of Solid
State/Condensed Matter Physics and
Chemical Dynamics. Specific topics with a brief description are given below in no particular order. |
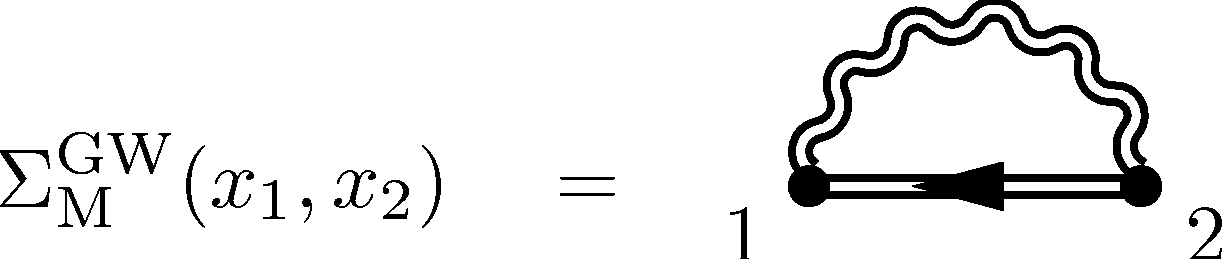 |
Many Body Perturbation TheoryThe Greens function approach focuses on the behavior of one (or a few) particle(s) embedded in the rest of the system. The single particle Greens function is a function of only two space-time variables yet contains enough information to construct the density matrix and total energy. The poles of this function yield exact single particle ionization and affinity levels, i.e. it is an exact independent particle like theory. Perturbation theories for the single particle (and higher particle number) Greens functions have been known for some time and so this approach is, in principle, systematically improveable. The focus on a few particles out of many makes these theories physically appealing. |
 |
Periodic SystemsInfinite systems have electronic properties that are absent in the finite sized systems that chemists are accustomed to concentrating their efforts in understanding. There are many features of periodic systems such as the presence of traveling waves, bands, fermi surfaces and long range ordering/short range localization that make their study interesting. In addition much of chemistry occurs in condensed phases or involve interfaces with such phases. There is also a richness in bonding in many complicated solid state systems of technological importance (such as metal oxides) that any chemist can admire. I am currenlty working on an implementation of periodic DFT in a Gaussian basis code that I hope to complete in the next year or so. I am also interested in the use of numerical atomic orbitals (NAOs) as a way to efficiently include Hartree-Fock exchange in plane wave DFT and implement wavefunction and perturbative methods. |
 |
Surface ScienceI am currently working on developing models to treat non-adiabatic dynamics for non-reactive scattering from metal surfaces. Previous work on NO/Au(111) by Sharoni Roy, Neil Shenvi and John Tully utilized a two state model and a non interacting dynamic model that was quite successful. The model Hamiltonian is taken to be the Anderson Hamiltonian which describes the coupling of a single localized orbital (a) with a band. The final term describes the fact that a second electron added to this localized state will increase the energy by a large coulomb interaction, U. The model of Sharani and Neil assumed an infinite U so that only one electron would ever inhabit the localized orbital. This is equivalent to a non-interacting spinless electron model in which a single electron would hop back and forth from the molecule to the metal and create electron hole pairs in the process. The hops are driven by the vibrating NO molecule which leads to energy transfer from molecular nuclear motion to electronic excitation. I am interested in extending this model to include multiple localized states and a finite U. This can be important in certain situations. One interesting example would be depolarization of a spin polarized beam of hydrogen atoms. The presence of a finite U requires a correlated (beyond mean field) description of the model Hamiltonian. This work is also in progress and it will be interesting to try different approaches to find approximate solutions to the Anderson model and see the effects of electron correlation on the ensuing dynamics. |
Hydrated ElectronHydrated electrons are generated when energetic particles (E > 7 eV) are absorbed or scattered by water molecules in aqueous solutions. The molecules are either fully ionized or excited, eventually leading to solvated, or hydrated, electrons. This species is ubiquitous and plays important roles in many fields. The formation, stabilization, relaxation dynamics, structure and spectroscopy of this species are all important topics.Previous work as a graduate student focused on improving the energetics of pseudopotentials used to describe the excess electron solvated by classical water molecules. Our work contributed to the understanding of the "blue-tail" of the characteristic absorption spectrum as well as the long standing and on-going debate over the assignment of binding motiffs in the observed photoelectron spectra of finite sized anionic water clusters. This debate has now nearly run its course though there is still work to be done on both the theoretical and computational sides. The absorption spectrum also deserves additional investigation, particularly on the dynamics in the blue tail as well as relations between the electronic structure of liquid water and the states of the excess electron. |
|
Fragmentation Schemes for Molecular LiquidsWave function methods developed in quantum chemistry have been very successful in the gas phase. These methods are systematically improveable and can be incredibly accurate for ground state properties. Due to poor scaling with system size these methods are difficult to apply to condensed phase systems. One solution for systems in which the electronic structure is localizable is to split the system into fragments, perform computations on the fragments and recouple the system in some way. Many such techniques exist (most of which are based on the many-body expansion) and we have constructed our own which we refer to as XPS.The most exciting aspect of these techniques is to apply them in to "on-the-fly" type approaches to molecular dynamics simulations. This will eventually allow parameter free, systematically improveable determination of equilibrium (density, structure, phase diagrams) and dynamic (diffusion, spectroscopy) properties of molecular liquids and solids. A challenge in this field is to apply these methodologies to excited states (ab-intiio exciton models?) and metallic systems. |