|
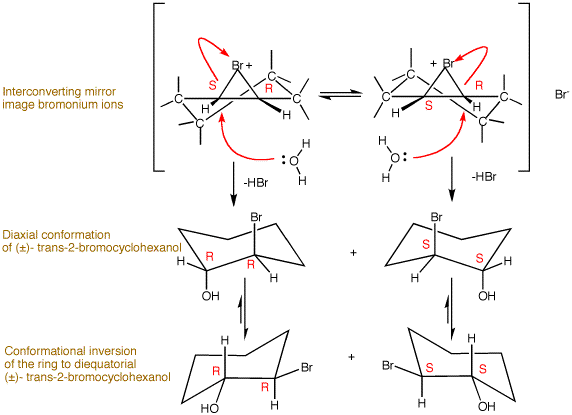
The halogenation of alkenes occurs by anti addition of diatomic halogen (X2) to an alkene through the intermediacy of a halonium ion in inert solvent. Review halogenation here. When this reaction is conducted in hydroxylic solvent (water, methanol and ethanol, typically), the solvent acts as the nucleophile to form halohydrins or, in the case of alcohols, halo ethers. Bromine is used in the following examples although chlorine or iodine may be used as well. The bromonium ion is a rapidly equilibrating mixture of mirror images (right panel). Water competes effectively with bromide ion as the nucleophile, not because it is a better nucleophile but because the bromide ion is highly solvated by water (less reactive) and there is much more water than bromide ion present. Water does an SN2 displacement at the C-Br bond that leads to a chair cyclohexane in the product. The mirror image transition states lead to mirror image diaxial bromohydrins. If water were to attack the bromonium ion at the alternate C-Br bond, a boat-like cyclohexane would occur through a transition state that is necessarily higher in energy than the chair-like transition state. The diaxial bromohydrins rapidly equilibrate to the more stable diequatorial conformers as a racemic mixture. |
 |
|
|
When a halohydrin is treated with a strong base aqueous KOH, methanolic sodium methoxide or ethanolic sodium ethoxide, proton exchange between the base and the halohydrin occurs reversibly and faster than intermolecular SN2 and E2 reactions (see left panel.) The bromohydrin alkoxide exists in two conformations: diequatorial and diaxial. While the former conformation is more populated, it is not suitable for an intramolecular SN2 reaction because the equatorial alkoxide cannot reach the rear of the C-Br bond to accomplish Walden inversion. If this contortional feat were possible, the epoxide formed would be trans-fused to the cyclohexane ring, an exceptionally strained ring! However, the less populated diaxial conformation is well-suited for rearside displacement to afford a cis-fused, stable epoxide. The observant student will notice that the achiral bromonium ion (vide supra) and the cis-fused epoxides are analogs of one another. They necessarily both have the configuration R,S. The first SN2 displacement occurs with water to give racemic (RR/SS) bromohydrin. The newly installed hydroxyl group, through the agency of its alkoxide acting as a nucleophile, inverts the remaining center to the R,S epoxide. This is an efficient reaction even though a strained ring is formed. The molar concentration of the alkoxide and the leaving group are high because they are proximate to one another with few degrees of freedom for random motion. |

|
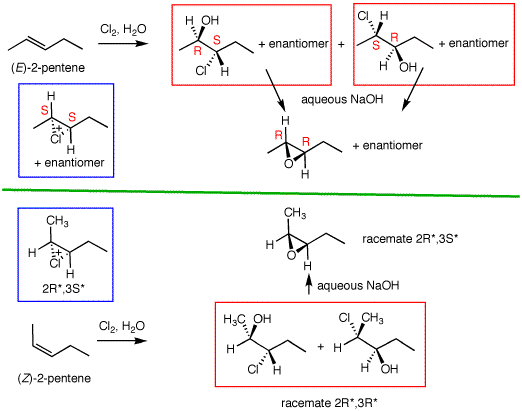
Consider the hydroxychlorination of (E)-2-pentene shown on the right. The racemic chloronium ion (blue box) can add water at two possible positions, C2 and C3, leading to the racemic 3-chloro-2-pentanol or the 2-chloro-3-pentanol. These structural isomers need not form in equal amounts. The 2-pentanol may be expected to be the predominate product owing to the less hindered attack of water near the methyl group as opposed to near the slightly larger ethyl group in the chloronium ion. There will to equal amounts of each enantiomer produced and the reaction will only occur by anti addition of the elements of HOCl. In spite of the formation of two structurally isomeric chlorohydrins, treatment of the mixture with base affords a single racemic epoxide. The ethyl and methyl groups were on opposite sides of the double bond; they are now on opposite sides of the oxirane ring by virtue of the double inversion. The R and S convention is used to designate the absolute stereochemistry of a single enantiomer. This method can also be used to designate relative stereochemistry. Consider the enantiomer (2R,3S)-3-chloro-2-pentanol and its enantiomer (2S,3R)-3-chloro-2-pentanol. To designate the racemate and its relative stereochemistry select the carbon bearing the lowest number assignment (C-2) in theenantiomer with the R-configuration and assign it as R*. The subsequent asymmetric carbons follow in kind. Thus, the racemate is (2R*,3S*)-3-chloro-2-pentanol. The racemic structural isomer would be (2R*,3S*)-2-chloro-3-pentanol. The racemic epoxide is designated (2R*,3R*)-3-ethyl-2-methyloxirane. |
|