|
The interrelated phenomena of hydrogen bonding and proton transfer represent two important aspects of molecular behavior. Hydrogen bonds are key features of molecular structure and reactivity. Low-barrier hydrogen bonds (LBHBs) are those characterized by great strength, short distance, and potential surfaces having either a very small barrier to proton migration or none at all. In the biochemical community, many believe the formation of very strong hydrogen bonds can supply the necessary stabilization to facilitate difficult reactions such as those observed to take place during enzymic catalysis. Our research efforts are an attempt to provide the first experimental evidence of low-barrier hydrogen bonding in a neutral molecular system. Malonaldehyde has long been the prototypical system for the study of intramolecular proton-transfer dynamics. Malonaldehyde represents the reaction center of many larger systems exhibiting interesting photochemical behavior, and the manifestation of an LBHB would have relevance to a wide number of potential applications. In the ground state it exists as a planar, intramolecularly hydrogen-bonded form with two equivalent asymmetric equilibrium configurations (Figure 1). The corresponding potential is a symmetric double well: two minima separated by a finite barrier. Rapid interconversion between the two configurations takes place by means of quantum mechanical tunneling, thereby leading to doubling of all rovibrational features. 
Figure 1. The tunneling coordinate for proton transfer in the groud electronic state of malonaldehyde. The majority of literature about proton transfer in molecular systems has dealt with the ground electronic state, and comparatively little is known about this process in the excited state. A characteristic of most excited-state proton transfers is the short time scales involved, often femtoseconds to picoseconds. The π* ← π (B1B2-X1A1) absorption feature of malonaldehyde resides in the vicinity of 266 nm and exhibits a large oscillator strength (akin to Figure 2). Theoretical predictions of LBHB phenomena in the B1B2 potential surface provide incentive for further experimental investigations of malonaldehyde. For the π* ← π state, the barrier to proton transfer has been predicted to disappear altogether, implying the existence of a single minimum potential well corresponding to a psuedo-cyclic structure in which the hydrogen is centered between the two oxygens. Resonance Raman spectroscopy (RRS) has been chosen as our method of interrogation of the π* ← π transition because it offers a means of extracting quantitative information from excited electronic states that are subject to rapid nonradiative relaxation processes. 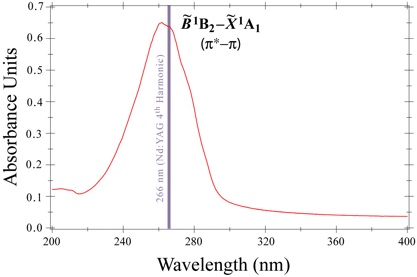
Figure 2. Near-ultraviolet linear absorption spectrum of acetylacetone, showing the strong π* ← π electronic band system. Preliminary attempts to employ RRS as a probe of excited state structure and dynamics have been focused on acetylacetone (AA), the enolic form of which represents a dimethyl substitution of the malonaldehyde framework (CH3(C=O)CH=(C-OH)CH3). Gas phase Raman spectra of this compound have implicated a LBHB in the electronically-excited B1B2 (π* π) manifold. In order to explore the influence of substituents upon LBHB phenomena, RRS has also been enlisted to interrogate the excited state structure and dynamics of hexafluoroacetylacetone (HFAA), where methyl groups in AA have been replaced by electron-withdrawing fluoromethyl moieties known to adversely affect the hydrogen-bond strength. The AA and HFAA Raman work provides strong evidence that comparable studies will be possible for malonaldehyde and might be extended to tropolone, which is another prototypical system for the investigation of intramolecular proton transfer and hydrogen bonding |