Index:
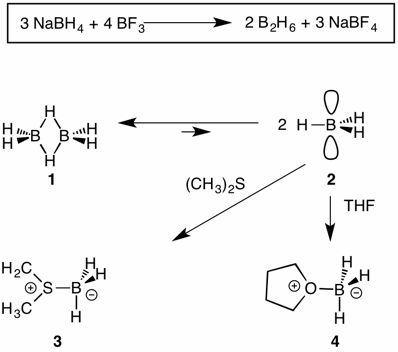
Hydroboration involves principally the reaction of borane (borine; BH3) with alkenes and alkynes. The use of metal hydrides for the reduction of boron trifluoride was developed by H. C. Brown, which led to the formation of diborane 1. Diborane is much more stable than borane 2 in the gas phase. In solvents that are Lewis bases, the monomer BH3, a Lewis acid, is trapped as a salt. The complexes formed with dimethyl sulfide (3) and tetrahydrofuran (4) are commercially available.
|
The addition of borane to an alkene (alkyne) is a reduction. The alkyl boranes formed by the addition of borane to alkenes are, for the most part, not the desired products. Oxidation of the alkyl borane with alkaline hydrogen peroxide affords an alcohol. Based upon the Pauling electronegativity boron (1.8) is less electronegative than hydrogen (2.2) which is less electronegative than oxygen (3.4). Whereas one of the carbons of ethylene 5 may be considered δ- and the other one δ+, in triethylborane both carbons are partial negative relative to their respective, covalently bound boron and hydrogen. This change constitutes a reduction of the carbon framework. When triethylborane is converted to ethanol 7, the carbon that was attached to boron in 5, becomes partial positive relative to oxygen. The overall process is a way to add water to a double bond.
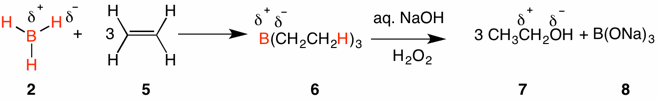
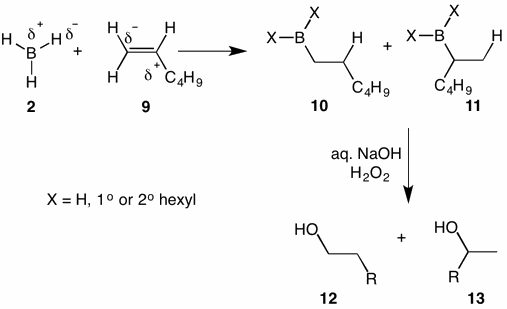
Hydroboration of terminal alkenes such as 1-hexene (9) provide 1-hexanol (12) and 2-hexanol (13) in ~90/10 ratio, respectively. The polarity of the double bond in 1-hexene has greater positive charge at the carbon bearing the alkyl group in the transition state for hydroboration. [Think about the stability of carbocations: 3o>2o>1o.] Excess borane favors the formation of boranes 10 and 11 (X=H). Such a reaction is optimized by addition of the alkene to the borane and not vice versa. Under these conditions one may assume that the ratio 10/11 is the same as 12/13. That is, 10 ---> 12 and 11 ---> 13. When an alkene/borane ratio of 3/1, i.e., stoichiometric, the ratio of 12/13 is 94/6, respectively. Terminal alkenes are relatively insensitive to substitution patterns. 1-Hexene and 3,3-dimethyl-1-hexene both give a 94/6 ratio of 1o/2o alcohols, respectively.
The overall process of hydroboration/oxidation is referred to as an anti-Markovnikov addition of water to a double bond. True enough! But the hydroboration step is not anti-Markovnikov but rather Markovnikov because boron is the electropositive element, not hydrogen. What causes the oxidation step to switch from Markovnikov to anti-Markovnikov is the replacement of boron with oxygen, which is more electronegative than hydrogen and boron.
Hydroboration of 2-methyl-2-butene (14) follows the regiochemical rules established above. Although some of the minor monoalkyl borane may be formed, only the major addition product 15 will be considered. Borane 15, owing to its branched structure, is highly regioselective when dialkyl borane 16 is formed. Dialkyl borane 16 does not react with a third equivalent of alkene at 0o to 25oC because of the steric bulk of the alkyl groups. Dialkyl borane 16 is a mixture of racemate and meso compounds. It is known as diisoamyl borane or disiamyl borane. It is often used as a reagent that reacts regioselectively with terminal alkenes, which are less hindered than alkene 14. The primary alcohol formed by the oxidation of such a trialkyl borane must be able to be separated from the 3-methyl-2-butanol that is also formed.
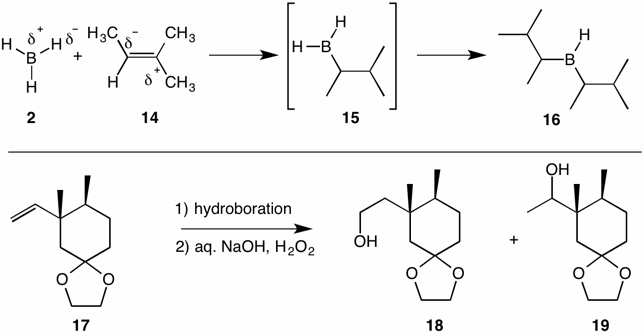
Alkene 17 has a double bond that is monosubstituted with a bulky alkyl group that is akin to a tert-butyl group. Hydroboration with borane 2 gave principally the desired primary alcohol 18 contaminated with secondary alcohol 19. When disiamyl borane 16 was employed, the primary alcohol was the sole detectable product after oxidation. Duringthe transition state for hydroboration the bulky portion of the substrate remains distant from the bulky portion of the reagent.
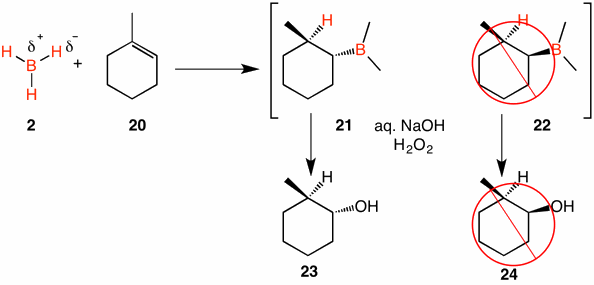
1-Methylcyclohexene (20) is achiral and has a trisubstituted double bond. Addition of borane in the Markovnikov sense would be expected to form racemic alkyl boranes 21 and 22. [Note: We are not concerned here as to how many B-H bonds react. Only one C-B bond is shown.] However, only alcohol 23 is formed! This result demonstrates that the boron and hydrogen atoms are added to the same side of the double bond. Moreover, the boron is replaced by oxygen with retention of stereochemistry.
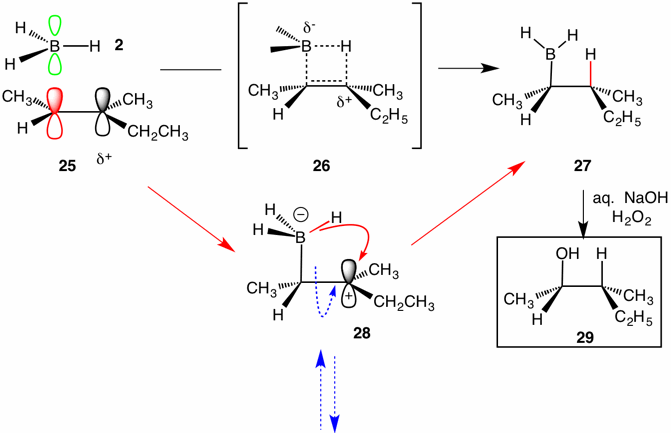
| 1-Methylcyclohexene (20) has a trisubstituted double bond constrained in a ring. Insight into the stereochemistry and mechanism of the hydroboration process can be attained by studying a pair of diastereomeric trisubstituted alkenes. Consider the hydroboration/oxidation of (E)-3-methyl-2-pentene (25) and the hydroboration/oxidation of (Z)-3-methyl-2-pentene (32). Ignoring any minor anti-Markovnikov products, cis addition of borane 2 to (E)-alkene 25 would provide alkyl borane 27 whereas the (Z)-alkene 32 would yield alkyl borane 34, the diastereomer of alkyl borane 27, both of which are racemic. When each of a pair of stereoisomers affords a unique stereoisomeric product, such a reaction is said to be stereospecific. Recall the bromination of (E)- and (Z)-2-butene. (E) --> meso dibromide; (Z) --> d.l-dibromide. The stereospecificity is controlled by mechanism. So how does hydroboration proceed? Is the addition of borane stepwise (28,30) or concerted (26,33)? A stepwise mechanism requires that rotation (---->) about the C-C bond in zwitterions 28 and 30 be much slower than transfer (____>) of hydride. If bond rotation were faster than hydride transfer, then both alkyl boranes would give in the same ratio from either alkene and subsequently the same ratio of alcohols 29 and 31. Since bond rotation is known to be very rapid, clearly another mechanism option must be considered, i. e., a non-stepwise mechanism ---namely, concerted bond formation and cleavage. Transition states 26 and 33 reflect the formation of the C-B and C-H bonds at the same time that the B-H bond is broken. |
|
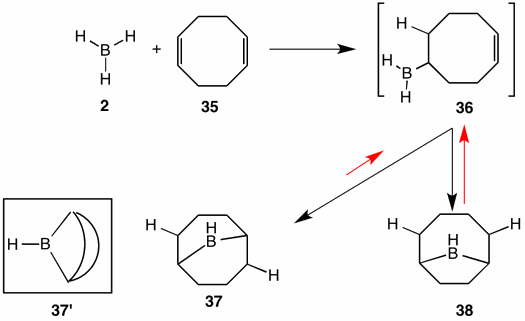
Hydroboration of 1,5-cyclooctadiene (35) proceeds in the expected manner at low temperature to provide unsaturated borane 36. An intramolecular hydroboration occurs faster than intermolecular hydroboration of diene 35. There are two constitutionsl isomers that are formed, --- namely, 9-borabicyclo[3.3.1]nonane (37) and 9-borabicyclo[4.2.1]nonane (38). Following the black arrows, these two bicyclics are formed under kinetically-controlled conditions. When the mixture is heated in tetrahydrofuran (THF), the red arrows now come into play as the bicyclics are in equilibrium with monocyclic borane 36. Under these conditions of thermodynamic control, the less stable bicyclic 38 is converted into the more stable isomer 37. This borane, often referred to as "banana borane" 37', is used for the regioselective hydroboration of other alkenes and alkynes.
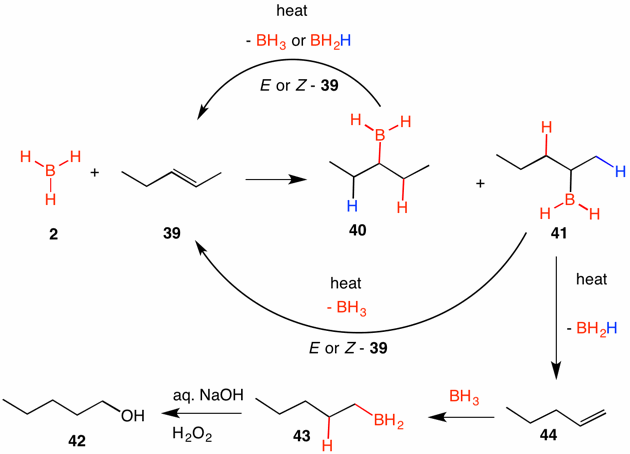
The reversibility of hydroboration manifests itself in the hydroboration of 1,2-disubstituted, straight chain alkenes at elevated temperatures. Consider the hydroboration of (E)-2-pentene, or (Z)-2-pentene for that matter, at low temperature (kinetic conditions). Two products, alkyl boranes 40 and 41, are expected with the latter in excess owing to steric considerations. [Note: Only monoalkyl boranes are shown to facilitate the discussion.] At higher temperature the two alkyl boranes are reversible. Alkyl borane 40 can have either the blue or red hydrogen participate in the elimination to (E)- or (Z)-2-pentene, which can undergo hydroboration to alkyl boranes 40 and 41 once again. Reversal of alkyl borane 41 gives either the 2-pentenes or 1-pentene 44. This alkene reacts irreversibly to form the stable, primary alkyl borane 43. The terminal borane is more stable than the internal boranes because the boron has two geminal C-H bonds with which to hyperconjugate while the internal boranes have a one C-H bond for hyperconjugation. Oxidation of the borane 43 affords 1-pentanol 42. The method may be applied to other unbranched chains. Imagine a mixture of all of the n-octenes. Under the conditions described all of them would yield 1-octanol. Accordingly, their action is known as a "zipper reaction" because the boron atom migrates down the carbon chain.
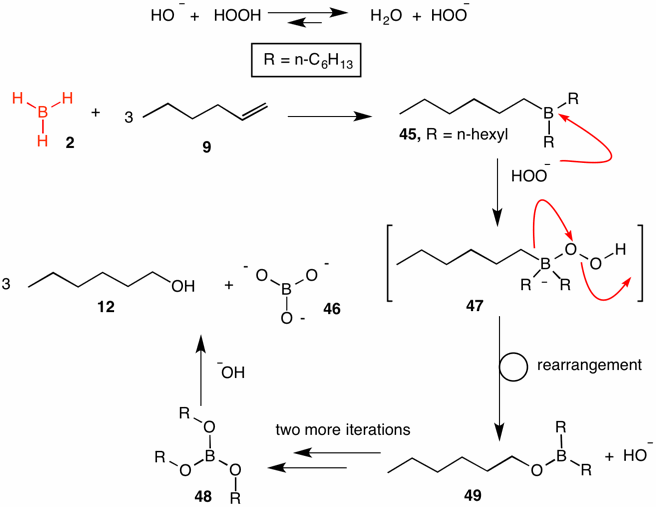
The standard method for oxidation of alkyl boranes to alcohols employs hydrogen peroxide in aqueous sodium hydroxide. Hydrogen peroxide is more acidic than water; consequently, the equilibrium lies to the right. Hydroboration of 1-hexene (9) leads to tri-n-hexyl borane 45. Boranes are Lewis acids. Addition of hydroperoxide anion to the alkyl borane provides anion 47. The pair of electrons in the C-B bond migrate from boron to the oxygen attached to boron. [The migration of the electron pair occurs without inversion, accounting for the retention of configuration.] This event is accompanied by the loss of hydroxide to yield alkoxy borane 49. The procedure amounts to the interpolation of oxygen in the C-B bond (C-O-B). The oxidation occurs twice more to afford tri-n-hexylborate (48). Hydroxide adds to the boron atom of ester 48 and exchanges hydroxide for alkoxide with protonation by water of the alkoxide to provide 1-hexanol (12).
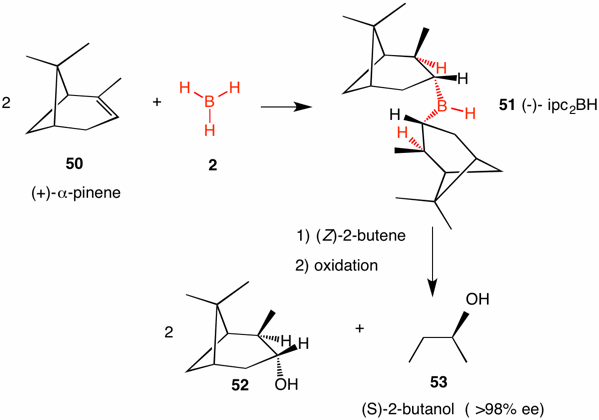
Hydroboration of optically active α-pinene (50) occurs twice to form diisopinocampheylborane [(-)-ipc2BH, 51]. This substance is a solid that is sensitive to moisture and oxygen. Borane adds to the double bond in the usual Markovnikov fashion. However, addition occurs on the less hindered face, ---namely, the face that is not obstructed by the proximate gem-dimethyl group. Hydroboration of symmetrical (Z)-1,2-disubstituted alkenes undergo asymmetric hydroboration. A typical example is the asymmetric hydroboration of (Z)-2-butene to afford after oxidation (S)-2-butanol (53) in greater than 98% enantiomeric excess and (-)-isopinocampheol (52).
March 14, 2016 (π-day, 3/14/16)