Chem 220 - Organic
Chemistry
Problem Set 8, Solution
Set
Chapter 9, Alkynes
Due: November 9, 2009

Friedrich Wöhler
(1800-1884)
(Wöhler possessed a wry sense of
humor) 1
2
|
Connections
Aluminum was once a precious metal
although it was plentiful. The problem was how to remove
it from its ore. Friedrich
Wöhler, of urea synthesis
fame, was able to accomplish this feat but by an
impractical method. He was to meet a young chemist,
Frank
Jewett, recently arrived in
Göttingen from Yale. Aware of the difficulty
Wöhler had had and probably encouraged by
Wöhler, Jewett, as a professor at Oberlin College,
passed the problem onto Charles
Martin Hall, a young student
at the college. Hall solved the problem in his family
garage. Thus was born Alcoa. At the same time in Spray,
North Carolina, Thomas
Willson, a Canadian, and
American James Moorhead were unsuccessfully trying to
refine aluminum using an electric arc. Unsuccessful in
purifying aluminum, they sought calcium metal. Heating
coal tar and lime in an electric furnace they obtained a
brittle material that produced a combustible gas upon
exposure to water. The material was not calcium nor was
the gas hydrogen. The pair was calcium carbide and
acetylene, the basis for Union Carbide (RIP).
|

Charles Martin Hall
(1863-1914)
|
The alkyne module in ORGO
gives a good review of acetylene chemistry.
|
1. Determine the structures A-K.
Explain your
reasoning.
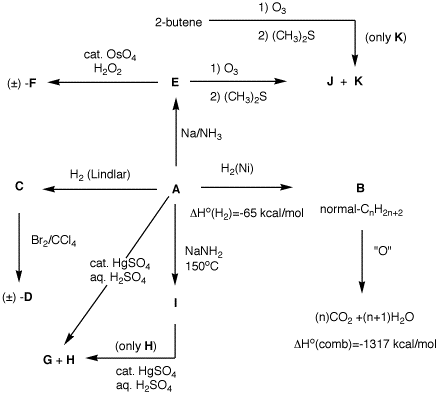
Structure A seems to be the center of
action. From its reactions it appears to be an alkyne. How
do we learn about its structure? Hydrogenation over a nickel
catalyst affords a straight chain alkane
B (normal chain). The heat of
hydrogenation is too much for a double bond. Using 3-hexyne
and 1-hexyne as models for internal and terminal alkynes in
the heats
of formation
table, the
respective heats of the respective heats of hydrogenation
are -65.5 and -69.2 kcal/mol for the two alkynes. The heat
of formation of n-hexane is -39.9 kcal/mol, the product of
hydrogenation. Clearly, A looks like an
internal alkyne. The heat of combustion of this n-alkane
gives a clue to the chain length. Compute the heat of
combustion of n-hexane (C6H14) from
the elements: 6 C (-94.05 kcal/mol) + 7H2 (-68.3
kcal/mol) = -1042 kcal/mol. The heat of combustion of
n-hexane is -1042 - (-40 (ΔHfo))
= -1002 kcal/mol. This value is less negative than -1317
kcal/mol by -315 kcal/mol. The heat of combustion for an
unstrained -CH2- group is -157 kcal/mol. The
value -315 kcal/mol accounts for two more -CH2-
groups. Thus, B is n-octane. A is
either 2-, 3- or 4-octyne. Na/NH3 reduction of
A gives E, which
contains an (E)-double bond. Ozonolysis of
E gives two products. This eliminate a
symmetrical alkyne for A and
symmetrical alkene for E.
K is acetaldehyde since it is the only
product of ozonolysis of 2-butene. Therefore,
J is hexanal (C6 straight
chain aldehyde). A is 2-octyne;
E is (E)-2-octene. Syn, 1,2-addition of
two hydroxyl groups to E gives
F [(2R*, 3R*)-2,3-octanediol]
as a racemate]. Lindlar reduction of
A affords C
[(Z)-2-octene]. C upon anti
addition of Br2 gives D,
(±)-(2R*, 3R*)-2,3-dibromooctane. NaNH2
isomerization of A forms I (1-octyne).
Hydration of the terminal alkyne I
gives only H, 2-octanone,
which also forms along with G
(3-octanone) upon hydration of A.
|
Note: There is another
way to find the number of carbons and hydrogens in B:
ΔHo(comb) = ΔHo(comb Cn)
+ ΔHo(comb Hn+1) -ΔHfo
ΔHo(comb) = nC + (n+1)H - 5n
(The term 5n is -5 kcal/mol/CH2 where n = #
carbons.) Now,
-1317 = -94.05n + [(n+1)(-68.3)] - (-5n)
-1249 = -157n
n = 7.9 or C = 8.
|
|
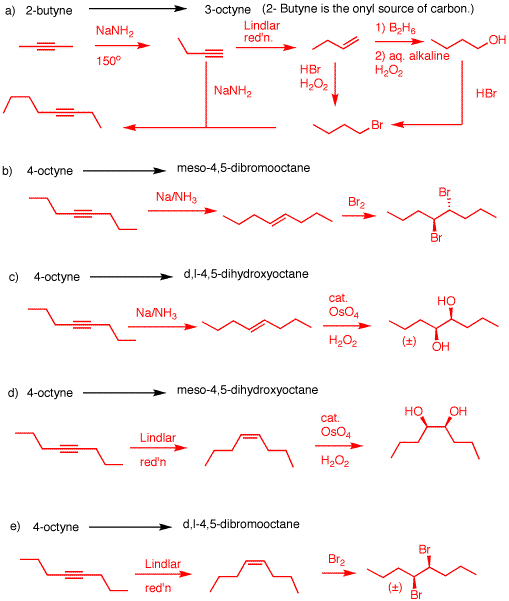
2. Provide reagents for the following
reactions. Explain your reasoning.
a) 4 + 4 = 8! This
is an alkyne alkylation. Isomerize 2-butyne to 1-butyne.
Split the pot of 1-butyne. Use one portion of 1-butyne to
form 1-bromobutane by either of the paths shown. Form the
anion of 1-butyne and alkylate with 1-bromobutane. When
1-butyne is formed as its sodium salt in the first step, you
can save 1/2 as the anion in preparation for the
alkylation.
b) You want meso. Anti addition of bromine to an (E)-double
bond.
c) You want a racemate. Syn addition of OsO4 to
an (E)-double bond.
d) You want meso. Syn addition of OsO4 to a
(Z)-double bond.
|
|
|
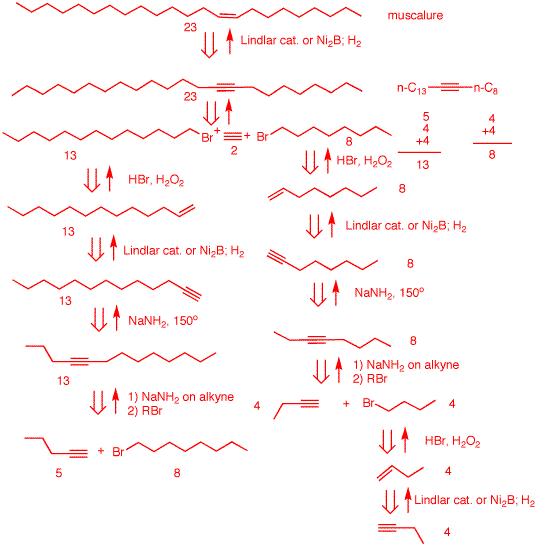
3. Design a synthesis of muscalure
[(Z)-tricos-9-ene], the sex attractant of the
common housefly, Musca domestica. As a source of
carbon you have available 1-butyne, 1-pentyne and acetylene.
You may use 1-pentyne and acetylene only once, i.e, only
seven of the carbons may be provided by these two alkynes.
All reagents are available.
Muscalure is a
C23 straight chain alkene with a (Z)-double bond
at C9. given the ground rules of the problem, use
a double acetylene alkylation along with syn reduction to
afford muscalure. One needs to prepare 1-bromotridecane
(C13) and 1-bromooctane: 13 = 5 + 4 +4; 8 = 4 +
4. {Note: actylene and 1-pentyne are used once. Follow the
retrosynthetic arrows to see how 1-bromooctane is prepared.
As in 2a, there is more than one way to convert a terminal
alkene to a primary bromide. 1-Bromooctane is also used in
the synthesis of 1-bromotridecane. How will you know if your
synthesis was a success? Flies will swarm about you.
|
|
|
4. Estimate the heat
of formation of 1-,2-,3- and
4-octyne. Equilibration of any one of these isomers with KOH
at 200oC produces about as much 2-octyne as
3-octyne both of which individually exceed the amount of
1-octyne. However, the amount of 4-octyne is less than the
amount of 2- or 3-octyne. Explain. [Hint: 2- and
3-octyne have an entropic advantage over 4-octyne.]
Using the heats of
formation table, one obtains the values for the three
hexynes. The estimate for 3-octyne an d 4-octyne can come
from 3-hexyne. The value for 3-decyne comes from
NIST
(National Institute of Standards and Technology) as do the
reported values. The correction is -5
kcal/mol/CH2 increase in chain length. Less
terminal alkyne is expected than internal alkyne because the
former have a higher heat of formation. There are two ways
to form 2- and 3-octyne but only one way to form the
symmetrical 4-octyne.
|
Alkyne
|
ΔHfo
Hexynes (kcal/mol)
|
Correction for Chain Length
(kcal/mol)
|
Estimated ΔHfo
for Octynes (kcal/mol)
|
Reported Values ΔHfo
(kcal/mol)
|
|
1-hexyne
|
+29.2
|
|
|
|
|
2-hexyne
|
+25.7
|
|
|
|
|
3-hexyne
|
+25.2
|
|
|
|
|
3-decyne
|
+5.2
|
|
|
|
|
1-octyne (from
1-hexyne)
|
|
-10
|
+19.2
|
+19.3
|
|
2-octyne (from
2-hexyne)
|
|
-10
|
+15.7
|
+15.2
|
|
3-octyne (from
3-hexyne)
|
|
-10
|
+15.2
|
?
|
|
3-octyne (from
3-decyne)
|
|
+10
|
+15.2
|
?
|
|
4-octyne (from
3-hexyne)
|
|
-10
|
+15.2
|
+14.4
|
|
|
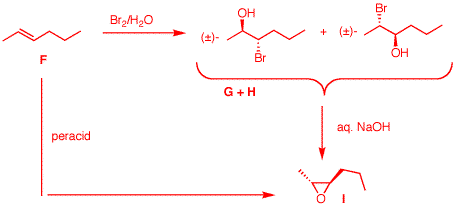
5. Two bottles are found on a laboratory
shelf labeled "alkyne A" and "alkyne B".
Hydrogenation of A or B over a platinum
catalyst gives the same alkane C.
A
and B have the same connectivity of
carbon atoms. Compound A
reacts with H2 in the presence of Lindlar's
catalyst to form D.
A
forms a (Z)-double bond.
Compound D reacts with O3 to form a single
compound E, C3H6O.
D is
a symmetrically substituted double bond (single
monofunctional product) as is A a
symmetrically substituted triple bond.
E cannot be acetone because ozonolysis
would have produced it from 2,3-dimethyl-2-butene
(tetramethylethylene), which cannot be derived from an
alkyne. E must be propionaldehyde,
CH3CH2CHO. Therefore,
D is (Z)-3-hexene and
A is
3-hexyne. On the other hand,
compound B reacts with aq.
H2SO4 in the presence of
HgSO4 to give two ketones J and K.
B is either
1-hexyne or 2-hexyne. 1-Hexyne would give only 2-hexanone
upon Hg++-catalyzed hydration. 2-Hexyne would give 2- and
3-hexanone. These two ketones are J and
K; B is 2-hexyne.
Under the same conditions,
A gives only J.
Since
A, 3-hexyne, affords only
J under these conditions,
J must be 3-hexanone and
K is 2-hexanone.
Compound B also reacts
with Na/NH3 to give F,
(F
is (E)-2-hexene) which itself
reacts with Br2/H2O to give a pair of
constitutional isomers, G and H.
Anti addition of
the elements of HOBr gives G and
H. See
below. Treatment of either
G or H with aqueous NaOH gives the same
compound I, C6H12O, that is
also formed by the reaction of F with peracid.
Halohydrin
formation followed by base treatment gives an even number of
SN2 inversions of stereochemistry: two. Peracid
gives zero inversions. Therefore, the two reaction sequences
give the same trans epoxide
I. What are the
structures of A-K? Explain and illustrate. [Note:
G and H are not distinguished from one
another. Pay attention to stereochemistry.]
|
|
6. The reaction on the right, which was
conducted on three different cycloalkynes, was reported in
1985 by Suzanne Abrams and Angela Shaw of the National
Research Council of Canada. Rather than use NaNH2
as your text suggests, they used the lithium salt of
tetradeutero-1,3-diaminopropane in
tetradeutero-1,3-diaminopropane as a solvent at room
temperature.
a) Name the alkynes used in these
experiments. [Note: Not surprisingly, cyclooctyne was
found to be unstable to the reaction conditions.]
n = 10;
Cyclododecyne
n = 11; Cyclotridecyne
n = 13; Cyclopentadecyne
b) The base in this experiment is
formed by adding n-butyllithium to the solvent,
tetradeutero-1,3-diaminopropane. Use the pKa
table to explain why this is a sound way to prepare this
base.
n-Butyllithium is
the conjugate base of the "acid" butane, pKa = ~50.
1,3-Diaminopropane can be modeled by ethylamine, pKa = 35.
The amine is the weaker acid; it is deprotonated by
n-butyllithium.
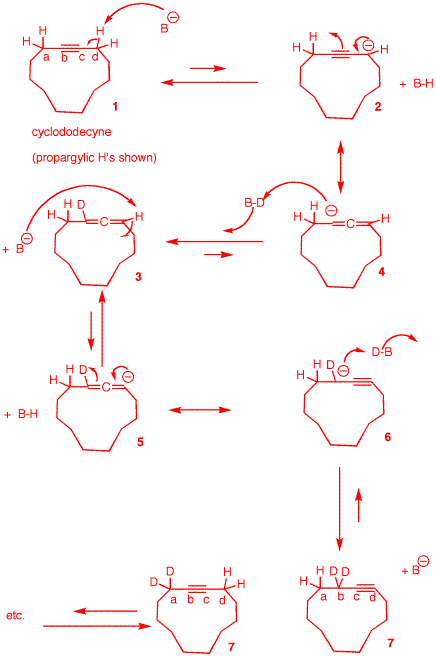
c) Provide an explanation (mechanism) as
to how each methylene group becomes deuterated. [Hint:
Such reactions are often called "zipper" reactions.
Why?]
If internal normal
alkynes can be isomerized to terminal alkynes, then the same
thing happens with cycloalkynes, except that there is no
irreversible last step. The final position of the triple
bond can be between any two contiguous carbons. Propargylic
hydrogens are to triple bonds what allylic hydrogens are to
double bonds. They are about the same pKa as the amine and
are reversibly deprotonated to form a propagylic anion
2 who resonance structure is allenyl
anion 4. [In this mechanism, the
amide base is B-; the deuterated amine is
B-D.] Deuteration of anion 4 leads to deutereated allene
3. Deprotonation of the vinyl hydrogen
of allene 3 affords allenyl anion
5 whose resonance structure is
deuterated propargyl anion 6.
Deuteration of this species provides dideuterated
cyclododecyne. Note that the position of the triple bond in
1 was between carbons a and b. In
7 the triple bond is between carbons c
and d. The triple bond "zippers" its way around te
ring.
|
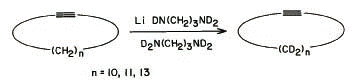 
|