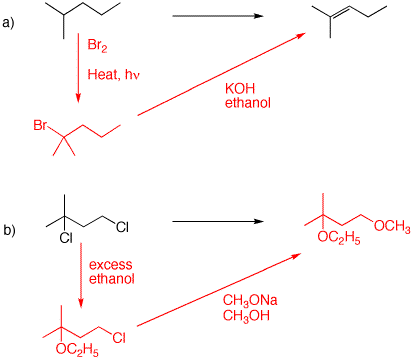
a) The question is: How do I introduce a double bond into a specific position in an alkane given what I know thus far? Since you know how to introduce halogen into an alkane and you know how to effect E2 elimination, this is a useful point of departure. A tertiary halide would be useful since it can undergo Zaitsev elimination leading to the more substituted alkene when a small base is employed. But which halogenation should be employed? Chlorination or bromination. The selectivity for chlorination is 1o(1):2o(4.5):3o(5.5) [Wade’s Text], while bromination is approximately 1o(1):2o(82):3o(1600) [See slide 12 in Radical.ppt., Anson, Fredricks, Tedder (1958)]. Clearly, bromination will be more selective for a tertiary hydrogen (~83%) than chlorination (~17%). This computation is in PS3 #1 and in slide 9 of Radical.ppt.
b) Tertiary halides are susceptible to solvolysis (SN 1) while primary halides are not! First treat the dichloride with an excess of ethanol to form the ethyl ether. Notice that no strong base (C2H5ONa) was used. This would cause substitution at the primary site (SN2) and E2 elimination at the teriary halide position [Cf. 4a]. Now use sodium methoxide/methanol to facilitate an SN2 substitution at the primary chloride. The existing ethyl ether is stable to these conditions. If this were not true, you couldn't prepare tertiary alkyl ethers by the Williamson ether synthesis.
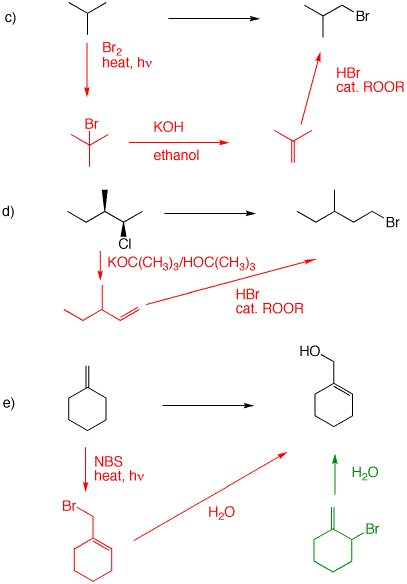
c) For the first step, see 4a. E2 elimination of HBr gives a single product, isobutylene. Peroxide-induced, anti-Markovnikov addition of HBr to the alkene affords isobutyl bromide (1-bromo-2-methylpropane). [If we haven't discussed "Markovnikov addition" at this point, look it up in your text.]
d) Formation of the bromide follows the protocol in 4c. How to obtain the alkene from the chloride? Use a bulky base (potassium t-butoxide) to minimize Zaitsev and maximize Hofmann E2 elimination.
e) Allylic C-H bonds have low Bond Dissociation Energies (BDEs). There are four, equivalent allylic C-H bonds in the starting material alkene. A resonance stabilized radical is formed which, in the second propagation step reaction with bromine, leads to the more substituted alkene - least substituted bromide. Some structural bromide may be formed (shown in green). Use the mixture in an SN1 solvolysis reaction with excess water, a reaction that proceeds through the resonance stabilized allylic cation. The role of NBS (N-bromosuccinimide) is to generate low concentrations of bromine, which favor radical reactions of bromine vs. addition to the double bond. The difference has to do with the kinetics of the two reactions. Radical bromination is first order in bromine; addition is second order in bromine. |
{kind=link}